Regulatory and strategic complications of marketing an oncology drug in Asia
The US is the most lucrative market for prescription drugs, routinely generating more than 50% of global sales in multiple therapy areas, including oncology. In the past five years, the Food and Drug Administration (FDA) has approved 88 innovator or new formulation drugs in oncology, few of which have made it into Japan and even fewer into China.
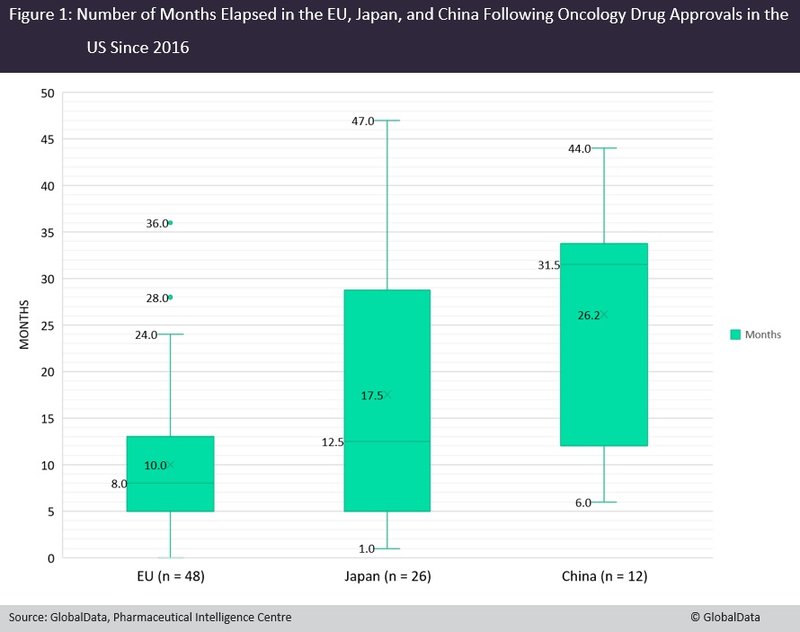
An analysis of GlobalData’s Pharma Intelligence Centre shows that 48 of these 88 agents have been approved in the European Union (EU), compared to 26 in Japan and 12 in China (Figure 1). While the median time for EU approval was eight months after US approval, the timeframe was much longer for Japan and China, at 12.5 months and 31.5 months respectively.
Some agents in the EU were approved more than 13 months after being approved in the US. These were outliers for a variety of reasons, including accelerated approval by the FDA, delayed European Medicines Agency (EMA) submission by the parent company, and targeting small indications with a low return on investment expected outside the US.
The approval of numerous drugs by the EMA within 12 months of application submission suggests that the main reason for delayed launch in the EU is the marketing company’s strategic decision to prioritise the US market rather than regulatory delays. This is unsurprising, as lower drug prices are the rule across the EU.
Japan and China offer unique challenges that could discourage smaller biotech companies from trying to enter these markets, especially without collaboration with a domestic partner. As a rule, both countries require the demonstration of the drug’s efficacy in an Asian population.
It is therefore necessary for companies considering these markets to either include domestic trial sites in their registrational trials or to launch a smaller, usually Phase II, study in Japan or China to satisfy the local regulatory agency.
While China’s National Medical Products Administration (NMPA) started accepting data from trials outside of China in 2018, the long timeframes associated with clinical trials meant that developers who had aimed for approval in China this year must have initiated a trial before 2018. As such, GlobalData expects a delay in utilising this regulation.
For companies with fewer resources, it makes sense to prioritise the US and EU markets and launch Asian trials only upon high confidence in their product’s chances for approval.
Of 12 products that were eventually approved in China, eight are manufactured by multinational pharma companies, and the remaining four by companies that struck a deal with a Chinese collaborator.
This suggests that for a small biotech company, unlike big pharma, collaboration is necessary, but not sufficient, for a swift entry into the China market.
Since 2016, Japan’s Pharmaceuticals and Medical Devices Agency (PMDA) has been able to review applications on a par with the EMA, while China’s NMPA is considered slower.
This means that the delayed launch in Japan compared to the US and EU is due to later submission because of the need for additional clinical data. While the NMPA has recently shown signs that it is reducing its time to review, the actual time-to-market for the few approved products still lags behind the US and EU.
When considering a lag time for launch in Japan and China, it is important to remember that a large portion of marketed products in the US will simply never make it into these markets, especially if they target niche patient populations.
Chinese biotech companies have started pursuing domestic approval first before expanding into other markets, as seen with Shanghai Junshi Biosciences’ Tuoyi (toripalimab) and Hutchison China MediTech’s Sulanda (surufatinib).
It is thus evident that both strategic and regulatory reasons drive the lag in time to market for novel agents in countries outside the US. The FDA’s accelerated approval programme has come under fire recently, but it is clear that it has contributed to the US being the leading country in patients’ access to innovative treatments, among other reasons.
For pharmaceutical industry data, comment and analysis, visit GlobalData's Pharmaceuticals Intelligence Centre.
Market Insight from